
What Drugs and Substances to Avoid to Lower Your Dementia Risk
I often get asked what people should avoid taking if they want to lower their risk of dementia. So this article works through the drugs and substances people use regularly, by prescription or otherwise, and asks which ones actually change the odds of cognitive decline and dementia, and how much we can trust each answer.
The shape of the evidence. Almost none of it comes from randomized clinical trials. You cannot ethically assign people to take a suspected harmful drug for years and wait to see who develops dementia. So most of what gets reported, including the alarming headlines, comes from observational studies, where the people who take a drug differ from the people who do not in ways that are hard to separate from the drug itself. A 2025 umbrella review in Molecular Psychiatry that pooled 68 meta-analyses across 11 drug categories said this plainly. The available data were primarily observational, confounding by indication and reverse causation were the dominant limitations, and randomized data were rare.
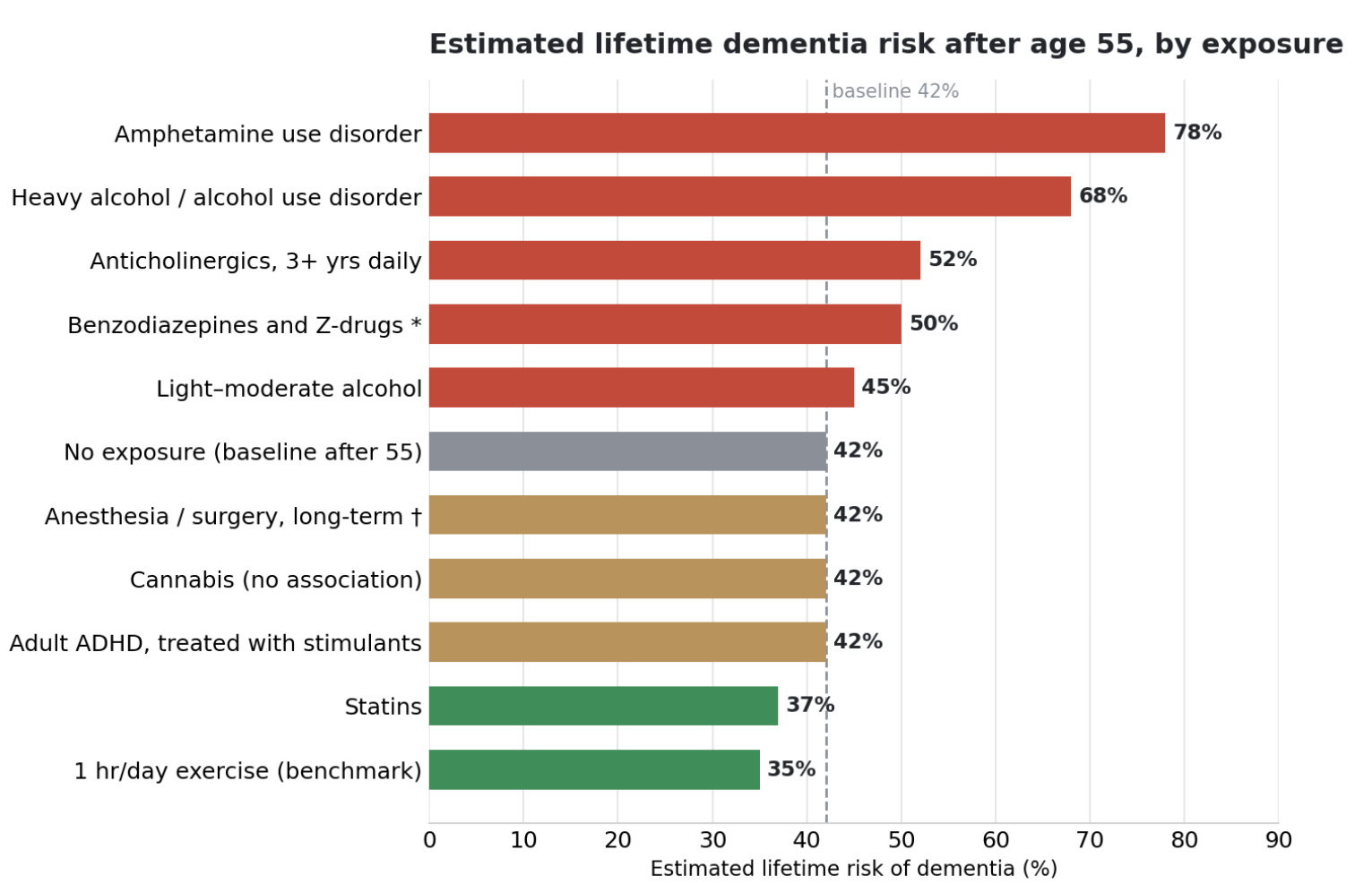
A note on the numbers. A phrase like “a 50 percent higher risk” is easy to misread, so throughout this article I have converted the hazard and odds ratios into approximate absolute lifetime risk, anchored to the 42 percent lifetime dementia risk after age 55 that Fang and colleagues reported in 2025. That figure comes from a large US community cohort, the Atherosclerosis Risk in Communities study, and represents cumulative risk from age 55 all the way to 95, with mortality counted as a competing event. It runs higher than older estimates of roughly 13 to 23 percent, partly because dementia is now documented more thoroughly and partly because most of the risk accrues after 85, so someone who does not reach their late 80s carries a much lower realized risk. For women and APOE4 carriers the baseline is higher still, around 45 to 60 percent. These converted numbers are illustrative rather than individual predictions, since the underlying studies span different populations and designs, but they make the size of each effect easier to feel. I also included one lifestyle benchmark, about an hour a day of exercise, so the drug effects can be read against a familiar comparison, with the caveat that the exercise estimate is observational and partly inflated by reverse causation. The chart below shows where each exposure lands.
With that backdrop, here is how the major players sort out, from the strongest signal of harm to the most reassuring.
Anticholinergic drugs carry the strongest and most consistent signal
What they are. These are medications that block acetylcholine, a chemical messenger the brain relies on for memory and attention, and they include some very common ones, such as the allergy and sleep aid diphenhydramine (Benadryl), the overactive-bladder drug oxybutynin, and the antidepressant amitriptyline. The signal here is the most consistent of any drug category, with a clear dose-response pattern and a plausible mechanism, even though it remains observational rather than trial-proven.
The evidence. The landmark prospective work, Gray and colleagues in 2015, found that cumulative use of strong anticholinergics for three or more years was associated with a meaningfully higher rate of dementia, with first-generation antihistamines, tricyclic antidepressants, and bladder antimuscarinics making up most of that exposure. A much larger 2019 nested case-control study by Dr. Carol Coupland and colleagues, covering nearly 285,000 people, narrowed the per-class signal to anticholinergic antidepressants such as amitriptyline and paroxetine, antipsychotics, antiparkinson drugs, antiepileptics, and the bladder antimuscarinics, where the odds ratio reached 1.65, close to the antipsychotics. A 2024 study of overactive-bladder drugs localized that further to oxybutynin, solifenacin, and tolterodine, while several other bladder agents showed no significant association. At cumulative exposure equivalent to taking a strong anticholinergic daily for at least three years, the odds of dementia rose by roughly 50 percent, which on the 42 percent baseline works out to about a 52 percent lifetime risk, the single class-level figure the chart uses. The 2025 umbrella review rated anticholinergics as moderate-certainty evidence of increased risk, the highest grade any harm-associated category received.
The gray-zone classes. Some everyday anticholinergics are harder to place. First-generation antihistamines like diphenhydramine and gut antispasmodics like dicyclomine are strongly anticholinergic and add to the cumulative load Gray measured, but neither reached its own significant dementia association in Coupland, so the case for limiting them rests on total burden rather than a standalone signal.
Better swaps exist for most of these. For overactive bladder, the beta-3 agonist mirabegron sidesteps the anticholinergic class. For allergies, the second-generation antihistamines cross into the brain far less. For mood, several modern antidepressants carry little anticholinergic activity.
Benzodiazepines show a signal of uncertain cause
Benzodiazepines, the class that includes alprazolam, lorazepam, and diazepam, are a partial exception to the no-trial rule, though not in the way the headlines suggest. Randomized trials do show that they impair memory and attention while a person is taking them, and discontinuation studies show that some of that recovers after stopping. What no trial has shown is that they cause dementia. That specific leap rests on observational meta-analyses, which report odds ratios in the range of about 1.4 to 1.8 and would, taken at face value, move lifetime risk from 42 percent to somewhere around 50 to 57 percent. The problem is that anxiety and insomnia, the reasons these drugs get prescribed, are themselves early symptoms of the dementia process, so the drug often gets started in people who are already on the path. A 2025 analysis was direct about this, finding only a modest increase in high-dose users and concluding that the association appeared driven by confounding from higher background rates of diabetes, cardiovascular disease, depression, and anxiety among users. The field has not resolved how much of the link is causal, so the true absolute number could lie anywhere between the baseline and the face-value figure. Caution about these drugs remains sensible on other grounds, including falls, short-term cognitive impairment during use, and dependence, which is why I steer away from them as sleep aids in my article on sleep and the brain.
Sleep aids split by type, and the over-the-counter ones are the bigger concern
Over-the-counter sleep aids are the ones I am most cautious about, because the active ingredient in most of them is an anticholinergic. The sedating antihistamine is usually diphenhydramine, the ingredient in Benadryl and ZzzQuil and the sleep component of combination pain-and-sleep products like Tylenol PM, Advil PM, and Aleve PM, or doxylamine, the one in Unisom SleepTabs. Both carry the cumulative anticholinergic concern described earlier, and prescribing guidance already lists them among the medications adults over 65 should avoid. For regular use, these are the sleep aids with the clearest signal to limit.
Z-drugs such as zolpidem (Ambien), eszopiclone (Lunesta), and zaleplon act on the same GABA system as benzodiazepines, and the dementia evidence looks much the same. A large cohort study found a 22 percent higher risk in users that fell to nothing once the underlying psychiatric and sleep conditions were accounted for, which points again to confounding by the sleep problem itself rather than the drug. There is also a mechanistic concern that these drugs blunt the deep-sleep stages that help the brain clear amyloid, so the reasonable reading is caution about long-term nightly use rather than alarm.
Trazodone, prescribed off-label at low doses for sleep, sits in a different and somewhat reassuring place. It is not anticholinergic, and although it is not proven to protect cognition, one cohort study found slower cognitive decline in users, possibly through its effect on slow-wave sleep, while a population study found no change in dementia risk in either direction. The evidence is mixed and modest, but nothing here points to the kind of harm the anticholinergic aids carry.
Orexin antagonists such as suvorexant (Belsomra) and lemborexant (Dayvigo) are the newest option and look mechanistically promising. They improve sleep without distorting its architecture, and early human work found that suvorexant acutely lowered amyloid and tau in the cerebrospinal fluid. This is preliminary rather than a dementia-prevention claim, but the direction is encouraging.
Melatonin shows no signal of cognitive harm and has a modest effect on sleep, though a 2025 preliminary analysis linked long-term use to higher heart failure risk. That analysis was observational and not yet peer-reviewed, and because melatonin is sold over the counter, supplement users may have been counted as non-users, so it raises a cardiovascular question rather than settling one.
A few feared drugs look neutral or protective
Statins, the cholesterol drugs such as atorvastatin and rosuvastatin, were once suspected of harming cognition, and many people still avoid them for that reason. The data now point the other way. If statins lower risk by something like a fifth, as parts of the 2025 umbrella review suggest, that moves lifetime risk from 42 percent down toward 37 percent rather than up.
Blood-pressure and metabolic drugs look protective in the same review, which found moderate-certainty evidence of reduced dementia risk for antihypertensives such as amlodipine and lisinopril, SGLT2 inhibitors such as empagliflozin and dapagliflozin, and GLP-1 receptor agonists such as semaglutide and liraglutide. The rare randomized data here supported treating high blood pressure as a way to lower dementia incidence.
Proton pump inhibitors, the acid-reducing drugs such as omeprazole and pantoprazole, generated dementia headlines a decade ago, but those associations have largely failed to hold up and are now regarded as likely confounded.
Menopausal hormone therapy moves women’s dementia risk in both directions depending on timing, and the older formulation may explain the late harm
This one does not fit on a simple harm-or-help axis, because the same drugs look protective or harmful depending on when a woman starts them. I lay out my own view on hormone therapy, including why I take it, in my article on sleep and the brain.
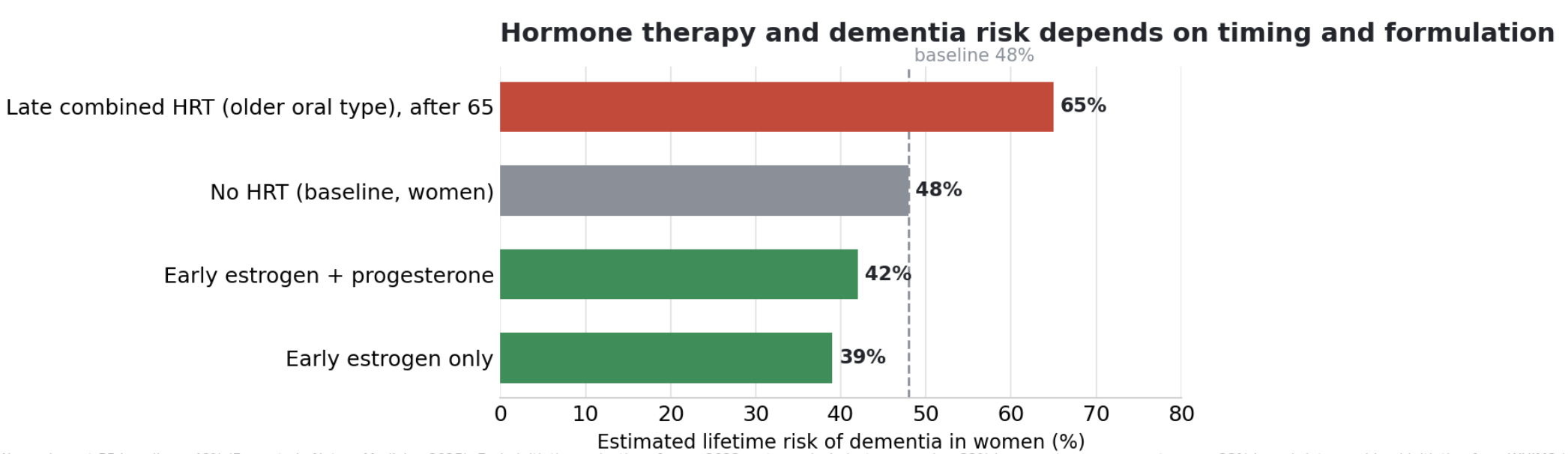
Started early, in the menopausal transition or the decade after it, hormone therapy is associated with lower dementia risk. A 2023 meta-analysis found that in midlife, estrogen-only therapy was associated with about a third lower dementia risk and estrogen plus progesterone about a quarter lower. On the roughly 48 percent baseline for women, that moves lifetime risk down to around 39 percent for estrogen alone and around 42 percent for the combination. The leading explanation is a critical window, in which estrogen supports neurons that are still healthy and a brain that still responds to it.
Started late, the picture flips, though the type of hormone may matter as much as the timing. In the Women’s Health Initiative Memory Study, the one large randomized trial here, oral conjugated equine estrogen plus medroxyprogesterone acetate, a synthetic progestin, begun in women aged 65 and older roughly doubled the rate of dementia, which on the women’s baseline corresponds to a lifetime risk near 65 percent. The estrogen-alone arm, without the synthetic progestin, showed only a non-significant trend, a hazard ratio of 1.49. Laboratory work suggests the synthetic progestin antagonizes estrogen’s neuroprotective effects while bioidentical progesterone does not, so the harm in older women may reflect both estrogen reaching neurons that are already compromised and the specific hormones that trial used.
The evidence gap is the part that often gets missed. The late-initiation harm signal comes almost entirely from that one older oral formulation. We do not have trials of the modern regimens many women use now, transdermal estradiol with micronized bioidentical progesterone, started in women over 65, with dementia as the outcome. The trials that did use those formulations enrolled recently menopausal women rather than the over-65 group. So what we can say is that we do not know whether starting a modern bioidentical regimen late carries the same risk, and the absence of data is not the same as evidence of safety.
The progestin question is a fair one, since estrogen alone looks better for the brain. The two concerns involve different organs. Estrogen stimulates the uterine lining to grow, and unopposed estrogen raises the risk of endometrial cancer, so a woman who still has her uterus needs a progestogen to keep that lining in check. A woman who has had a hysterectomy can take estrogen alone, which is why the estrogen-alone trial enrolled only hysterectomized women. The reconciliation with the dementia data is that the harm came from the synthetic progestin specifically, not from progesterone as a class, so modern micronized progesterone can provide the uterine protection without necessarily carrying the same brain effect.
The practical reading is that both timing and the type of hormone shape the risk, and the old headline from the late-initiation trial, which used an older oral formulation, does not necessarily describe early initiation or the modern bioidentical regimens.
Anesthesia and surgery deserve a careful answer, especially for APOE4 carriers
Two timeframes. This question comes up often, and the answer separates into two timeframes that get conflated.
In the short term, two things can happen after surgery. One is postoperative delirium, a fluctuating state of confusion in the days after an operation. The other is a measurable dip in memory and thinking that can last weeks to a few months. This short-term effect is a separate outcome from lifetime dementia, which is why the chart above places anesthesia only at its long-term value, the baseline. In absolute terms the short-term dip shows up in a sizable share of older surgical patients, on the order of one in six, and APOE4 carriers appear modestly more vulnerable to it. A meta-analysis of 22 studies and 6,734 patients found APOE4 associated with these short-term cognitive changes within one week, with an odds ratio of 1.89, and a smaller but present association at one to three months, an odds ratio of 1.67. Notably, that same analysis found the association faded by one year and was not significant for delirium itself.
In the long term, the picture is reassuring and the conflicting findings are themselves informative. A 2025 systematic review in BMC Geriatrics restricted to population-based cohort studies found no increase in dementia risk from general anesthesia, which usually relies on agents such as propofol and sevoflurane, or regional anesthesia, with a hazard ratio of 1.30 and a confidence interval that crossed one. A large cohort directly comparing general versus regional anesthesia found no difference. The pattern that runs through this literature is that positive signals tend to come from case-control designs while record-based prospective cohorts come back null, and an earlier meta-analysis showed the association appearing mainly when exposure was reconstructed from interviews rather than records. That pattern points toward confounding and recall bias rather than a durable anesthetic effect on the disease itself, so a person’s underlying lifetime risk looks essentially unchanged at the 42 percent baseline.
For an APOE4 carrier, the message is not to fear a medically necessary operation, because the risk of an untreated condition almost always outweighs a transient and reversible cognitive dip. For an elective procedure, there is room to plan. A patient can ask the anesthesiologist about the brain-health protocols that already exist through professional anesthesiology societies, ask about depth-of-anesthesia monitoring so sedation goes no deeper than needed, ask to minimize benzodiazepines and anticholinergics around the procedure, and ask whether a regional or nerve-block technique is feasible, mostly because it reduces opioid load and pain, both of which drive delirium. Establishing a brief cognitive baseline beforehand makes any later change interpretable rather than guessed at, and a recovery environment with daytime light, real sleep at night, family presence, and the early return of glasses and hearing aids lowers delirium risk.
The case for protective moderate drinking has not survived the genetic studies
The old J-curve. For decades the story was a J-shaped curve, with light drinkers appearing healthier than both abstainers and heavy drinkers. The best current evidence has taken that apart, and I went through it in depth in my earlier article on alcohol, brain health, and longevity, so here I will keep it to the dementia-relevant core.
The genetic evidence changed the picture. Mendelian randomization uses gene variants that influence how much people drink as a kind of natural experiment, sidestepping the lifestyle confounding that makes moderate drinkers look healthy for reasons unrelated to alcohol. The largest of these analyses, Topiwala and colleagues in 2025, drew on genetic data from more than two million people and found a roughly linear relationship between alcohol and dementia, with risk climbing from low levels and no protective sweet spot in the moderate range. The apparent benefit of light drinking in older observational work looks like an artifact of sick-quitter effects, healthy-user bias, and the social connection that tends to accompany moderate drinking.
Two caveats keep this accurate. The effect sizes from the genetic data are modest, on the order of a 15 percent increase in dementia risk per a few additional drinks a week, which on the 42 percent baseline lands near 45 percent and is smaller than carrying one APOE4 allele or having midlife hypertension. And Mendelian randomization has its own blind spot, because the gene variants that predict drinking also tag impulsivity and related behaviors, so part of the harm assigned to alcohol may reflect correlated traits. At the heavy end there is no ambiguity. A 2018 analysis in Lancet Public Health of more than a million French hospital admissions found that alcohol use disorders more than tripled dementia risk, which converts to a lifetime risk near 68 percent, and excessive alcohol now sits on the Lancet Commission list of modifiable risk factors. For APOE4 carriers there is suggestive evidence of amplified vulnerability, which I cover in the alcohol article.
Stimulant risk tracks the exposure and the population, not the drug class label
Stimulants are the category where the dementia data look most contradictory at first glance. Heavy illicit use, mostly methamphetamine, shows one of the steepest risk signals of anything in this article, while prescription stimulants taken for ADHD, such as methylphenidate and amphetamine salts, show none. It would be a mistake to read that as one drug class behaving two different ways, because amphetamine and methamphetamine sit in the same class. The split is about how much drug reaches the brain and who is taking it.
The steep signal comes from a nationwide cohort study in Taiwan, where people with amphetamine-related disorders had close to a fivefold higher rate of dementia after adjustment, which on the 42 percent baseline corresponds to a lifetime risk near 78 percent, spanning Alzheimer’s, vascular, and other types. That exposure is a use disorder, which in Taiwan is overwhelmingly methamphetamine, taken at doses far above any prescription, often smoked or injected for rapid high peaks, and frequently alongside the hyperthermia, sleep loss, and polydrug use that drive dopaminergic neurotoxicity. The methyl group on methamphetamine also makes it more lipophilic than prescription amphetamine, so it enters and accumulates in the brain more readily. On top of the pharmacology, a use-disorder population carries heavy independent dementia risk from tobacco, stroke, head injury, vascular disease, and social deprivation, so the fivefold figure is part drug and part everything that travels with it.
The treated population looks different in the data we have. A 2023 cohort study in JAMA Network Open of more than 109,000 adults found that an adult ADHD diagnosis was associated with a 2.77-fold higher dementia risk, near 67 percent in absolute terms, but that no increased risk appeared among those treated with prescription stimulants, who stayed close to the 42 percent baseline. That interpretation is not settled, because treated ADHD may differ in severity from untreated, and none of this is randomized. What it supports is the narrower claim, which is that supervised stimulant treatment at therapeutic doses does not carry the signal seen in heavy illicit use, rather than a clean statement that the drug class itself is safe.
Other street drugs vary by mechanism
Cocaine acts mainly through the blood vessels, and repeated small episodes of reduced blood flow plus accelerated arterial disease make vascular cognitive impairment the plausible long-term concern. The framing matters here, because the meta-analytic review of 46 studies I would point to measured cognitive performance in chronic users, finding moderate or larger deficits in attention, episodic memory, and working memory. That is a cross-sectional snapshot of current users, not an incidence study, so it does not yield a lifetime dementia risk the way the amphetamine and alcohol cohorts do. There is no reliable figure to convert. The long-term dementia risk from cocaine is mechanistically plausible through its vascular damage but has not been quantified in the same way.
Opioids, such as oxycodone and hydrocodone, appear to carry an elevated dementia association in cohort data, with proposed mechanisms including repeated low-oxygen episodes and effects on dopamine and hippocampal plasticity, though as with benzodiazepines the confounding from the conditions that lead to long-term opioid use is substantial, and as with cocaine the data do not support a firm lifetime-risk number.
Cannabis is the surprise. A 2026 study in BMJ Mental Health combining large UK and US cohorts with a Mendelian randomization analysis found no association between cannabis use and dementia or accelerated cognitive decline, and users actually performed modestly better on some cognitive tests, a finding the authors attribute partly to the higher education levels of recreational users rather than a true benefit. Acute intoxication and heavy use do impair short-term memory and executive function, and the authors were careful to say this does not make cannabis risk-free for other health outcomes. On the specific question of dementia, though, the signal is absent in the best current data.
Psychedelics, MDMA, and ketamine sit mostly outside the dementia evidence
Why they group together. They come up often, and the dementia question for all three rests on short-term cognitive studies in heavy users rather than on incidence data, so there are no lifetime-risk numbers to convert and none of them appear on the chart.
MDMA raises a brain-harm concern, but the evidence is not strong. Chronic heavy recreational use damages serotonin neurons in animal studies, and abstinent heavy users show deficits in episodic memory, working memory, and attention, along with hippocampal changes on imaging. These are observational comparisons of users with non-users, usually entangled with polydrug use, and they measure cognitive performance rather than dementia, so the signal is suggestive rather than established. The effect also looks dose-dependent. The deficits cluster in heavy, high-cumulative users, while light or occasional users show little or no measurable impairment, and the small effect that does turn up in current light users appears to fade after several months without the drug. The limited supervised dosing used in MDMA-assisted therapy is a far smaller exposure than chronic recreational use and has not been linked to lasting cognitive harm, though no study has followed any dose level out to dementia.
Ketamine splits by dose and pattern the same way the stimulants did. Frequent heavy recreational use, on the order of several times a week, is associated with memory impairment that appears to reverse after about a year of abstinence, along with bladder injury. Infrequent recreational use and the low therapeutic doses used for depression do not carry that signal, and ketamine is being studied as a neuroprotective agent rather than a harmful one.
Classic psychedelics point the other way entirely. Psilocybin and LSD are not known to be neurotoxic at typical doses, and the research interest is in possible benefit through activation of the 5-HT2A receptor, which drives neuroplasticity and the growth of new synaptic connections. A 2024 review laid out the case for psilocybin in Alzheimer’s on those grounds, and a recent case report described temporary functional gains in a woman with advanced disease, which I covered in my article on a single dose of psilocybin and earlier in magic mushrooms and longevity. None of this shows that psychedelics prevent dementia, and the human evidence is early, but nothing here points to raised risk.
The bottom line
So where does all of this leave us? A few clear conclusions hold up.
Cumulative anticholinergic burden is the part most people can act on, because the signal is consistent and most of these drugs have gentler substitutes that keep lifetime risk nearer the 42 percent baseline than the 52 percent that years of use implies. Here is the short list of what to avoid or ask about swapping:
Sedating antihistamines such as diphenhydramine (Benadryl), hydroxyzine (Atarax, Vistaril), and chlorpheniramine, including over-the-counter PM sleep aids like Tylenol PM and ZzzQuil, where trazodone, the orexin antagonist suvorexant, or melatonin are lower-risk choices for sleep
Tricyclic antidepressants such as amitriptyline, doxepin, and nortriptyline, along with paroxetine (Paxil)
Overactive-bladder antimuscarinics such as oxybutynin (Ditropan) and tolterodine (Detrol), where mirabegron is the lower-burden swap
Antispasmodics such as dicyclomine (Bentyl) and hyoscyamine, and the muscle relaxant cyclobenzaprine (Flexeril)
Some older antipsychotics and antinausea drugs such as chlorpromazine, promethazine (Phenergan), and meclizine
Heavy alcohol, where the lever is cutting back, since the genetic evidence shows harm climbing from low levels rather than a safe moderate zone
Illicit stimulants, mainly methamphetamine, along with cocaine, which sit at the severe end of the chart
The rest, in brief. Hormone therapy moves risk in either direction depending on when it starts and which formulation is used, lower with early initiation near menopause and higher with the older oral regimen begun in the mid-60s. Several of the most-feared everyday drugs, statins among them, trend toward lower risk. Surgery and anesthesia cause short-term and reversible cognitive effects that APOE4 carriers feel a bit more, but the durable dementia risk looks unchanged, and the leverage is in optimizing an elective procedure rather than avoiding a necessary one.
One caution. None of this is a reason to stop a prescribed medication on your own, and abrupt discontinuation of many of these drugs carries its own risks. This is general scientific information rather than individualized medical advice, and any change to a medication should be made with the clinician who prescribed it.
The next few years should bring direct evidence
Deprescribing trials are coming. As blood-based markers and brain-aging clocks get better at detecting risk early, I expect the next few years to bring deprescribing trials that finally test some of these associations directly, which is the evidence we have been missing. A study that randomizes long-term anticholinergic or benzodiazepine users to a structured taper and then tracks cognition and biomarkers over time would turn today’s observational signals into something closer to cause and effect. The same design applied to modern transdermal estradiol with micronized progesterone in women over 65 would close the formulation gap this article keeps returning to. At NeuroAge we are building toward exactly this kind of early detection, since knowing who is on a faster aging trajectory is what makes a prevention trial possible in the first place.

Written by
Dr. Christin Glorioso, MD PhD
Dr. Glorioso is the founder and CEO of NeuroAge Therapeutics. With her background in neuroscience and medicine, she is dedicated to revolutionizing brain health and helping people maintain cognitive vitality.
Learn more about Dr. Glorioso

