
A tau drug and an at-home amyloid injection point Alzheimer’s treatment toward combinations matched to each person’s biology (new study results)
The Alzheimer’s Drug Discovery Foundation flagged two developments in a single week, one on a tau drug and one on an amyloid drug, that together sketch out where Alzheimer’s treatment is heading. I follow the ADDF closely because it funds one of our studies at NeuroAge Therapeutics, the company I lead, so its read on the field reaches me early.
The foundation has been at this since 1998, when it was set up by Leonard A. and Ronald S. Lauder of the Estée Lauder cosmetics family, and it is the only public charity devoted entirely to funding Alzheimer’s drug development. Working through a venture philanthropy model, in which it backs drug programs the way an investor would rather than funding basic science alone, it has put nearly $400 million into more than 790 research programs across 21 countries, and it helped bring the first Alzheimer’s PET brain scan and the first blood test to market.
On July 14, 2026, Biogen presented phase 2 results for diranersen, a drug that lowers tau, one of the two proteins that define the disease. A day earlier, the FDA approved a version of the amyloid antibody Leqembi (lecanemab) that patients can start at home with a weekly injection under the skin instead of beginning with months of intravenous infusions. One announcement is about reaching a new target. The other is about making an existing treatment easier to give. Both matter for the same reason, which is that the field is shifting away from single drugs and toward combinations chosen for what a given person’s blood shows about their disease.
Alzheimer’s has two signature proteins, and until recently the approved drugs cleared only one of them. The first is amyloid-beta, a protein fragment that in Alzheimer’s clumps together into sticky deposits called plaques, which collect in the spaces between neurons. The second is tau, a protein that normally helps hold the internal scaffolding of a neuron in place. In Alzheimer’s, tau changes shape and twists into tangles inside the neurons themselves, and those tangles track more closely than the plaques with where and when brain cells are lost. The approved antibodies lecanemab and donanemab both work by clearing amyloid, and they slow decline modestly in people with early disease. Tau has been the harder target, which is part of why a tau drug producing any clinical signal is notable. I wrote about how the broader treatment landscape has been catching up to this two-protein biology in The Alzheimer’s Pipeline Is Finally Catching Up to the Biology.

The diranersen data are the first from a randomized trial where lowering tau moved both the biology and the clinical measures
Diranersen is an antisense oligonucleotide, which is a short piece of synthetic genetic material designed to bind the instructions a cell uses to build a protein and mark them for disposal. In this case it targets the messenger RNA for tau, so the cell makes less tau to begin with rather than trying to clear tangles after they form. It reduces both the tau inside neurons and the tau that spreads between them, across all forms of the protein. Because it does not cross from blood into the brain on its own, it is given intrathecally, meaning injected into the fluid around the spinal cord. This was first shown to lower tau in cerebrospinal fluid in a phase 1b trial published in Nature Medicine, with a later exploratory analysis in Nature Aging hinting at slower decline at higher doses.
The phase 2 CELIA study enrolled 416 people with early Alzheimer’s, none of whom had previously received an anti-amyloid drug, and it produced a robust biomarker effect. Across all doses tested, spinal-fluid total tau fell by roughly 50 to 65 percent from baseline, and tau measured by PET brain imaging decreased across every region examined. This makes diranersen the first tau-directed therapy to lower both spinal-fluid tau and brain tau on imaging across all doses in a phase 2 study. On the clinical side, the 60 mg dose given every six months showed the strongest response, with decline slowed by about 26 percent on the CDR-SB, a standard scale of cognition and daily function, along with 42 percent on a cognitive test called ADAS-Cog13 and 50 percent on the Mini-Mental State Examination.
The study did not meet its primary endpoint, and understanding why is where the interesting question sits. CELIA was built to test whether higher doses would produce greater clinical benefit, and that dose-response relationship did not appear. Every dose slowed decline relative to placebo, but the higher doses did not do better than the lower one, and the strongest clinical signal came from the lowest dose even though the higher doses lowered tau more. That divergence between the amount of tau removed and the amount of clinical benefit is the central puzzle Biogen carries into phase 3, and it raises a real question about how much tau reduction is optimal rather than simply how much is possible. The limits belong here as well. Most of the endpoint differences reached only nominal statistical significance, one measure of daily function did not separate from placebo, and this is a mid-stage result rather than a confirmatory one. As the ADDF put it in its statement on the data, these are the first randomized data to show a tau-lowering drug producing both a strong biomarker effect and a clinical signal, with the dose question now central to designing the next trial.
One practical point in diranersen’s favor is that its mechanism should avoid a specific safety problem of the amyloid antibodies. The anti-amyloid drugs carry a risk of amyloid-related imaging abnormalities, brain swelling or small bleeds visible on MRI that require monitoring. Because diranersen acts on tau rather than amyloid, that particular risk is not expected, and the CELIA safety data were consistent with that. Its own burden is different, since it involves repeated spinal injections.
A tau drug matters most in the diseases where tau is the whole problem and nothing is approved
Framing diranersen only as a future partner for amyloid drugs understates its value, because there are diseases driven by tau alone where a tau-lowering drug is the entire therapeutic strategy. These are the primary tauopathies, a group that includes frontotemporal dementia caused by mutations in the MAPT gene, progressive supranuclear palsy, and corticobasal degeneration. In these conditions tau accumulates and neurons are lost without amyloid playing the driving role that it does in Alzheimer’s, so the amyloid antibodies have nothing to act on. There are no approved treatments that change the course of any of them, which leaves a group of patients with a clear molecular cause and no drug aimed at it.
A drug that lowers tau at the source fits these diseases in a way it does not quite fit Alzheimer’s. Diranersen works by reducing production of the tau protein itself across all of its forms, and in frontotemporal dementia caused by a MAPT mutation, the problem is precisely that the gene produces abnormal tau. Lowering the output of that gene is close to addressing the cause directly rather than managing a downstream effect. The same reasoning extends to progressive supranuclear palsy and corticobasal degeneration, which are defined by tau aggregation and where tau-directed therapies are now moving into later-stage trials. A study of diranersen in corticobasal syndrome is planned at the University of California, San Francisco, with support from the Alzheimer’s Association, and separate tau therapies have advanced to phase 3 in progressive supranuclear palsy.
This changes how to read the Alzheimer’s result. The CELIA data matter for Alzheimer’s, but they also serve as human proof that lowering tau in the brain is achievable and can track with clinical change, which is the assumption the entire primary tauopathy field rests on. A tau drug that earns approval in Alzheimer’s would arrive with a safety and dosing record that other tauopathy programs could build on, and the patients with MAPT frontotemporal dementia or progressive supranuclear palsy, who currently have nothing, may have the most to gain from a tau-lowering approach even though Alzheimer’s is where the trials are largest.
The subcutaneous Leqembi approval is about logistics, and logistics are what make combinations realistic
The FDA’s decision lets patients begin lecanemab with a weekly injection under the skin rather than starting on intravenous infusions. An autoinjector delivers the dose in roughly 15 seconds once a week, compared with hour-long infusions at a hospital or infusion center. Under the prior approval, patients spent about 18 months on intravenous dosing before they could move to subcutaneous maintenance, so this is the first time treatment can start at home. New data presented at the same conference indicated the weekly subcutaneous start performs similarly to the intravenous regimen with a generally consistent safety profile. Lecanemab’s original benefit in early Alzheimer’s was established in the pivotal trial published in the New England Journal of Medicine.
This matters for combination therapy because a multi-drug, multi-year regimen only works if each drug is tolerable to live with. If the future of Alzheimer’s care involves clearing amyloid, lowering tau, and possibly addressing inflammation or metabolism over a span of years, then every component that requires a hospital visit compounds the load on patients and caregivers. Anything that moves a therapy toward at-home administration makes it easier to combine with others. Other companies are moving the same direction, with Eli Lilly testing a subcutaneous form of its investigational anti-amyloid antibody remternetug. The ADDF framed this in its statement on the approval as a step toward the kind of scalable care model the disease will need as treatment becomes long-term and multi-drug.
A central reason to measure tau is that anti-amyloid drugs work less well once tau is high
The clinically important point is not that patients fall into a tau camp and an amyloid camp, but that the amount of tau a person already carries changes how well an amyloid drug will work for them. In Alzheimer’s, amyloid generally accumulates first, and tau tends to follow and to track more tightly with where and when neurons are lost. The amyloid antibodies were tested in people who had both proteins, and their benefit was consistently larger in those who started treatment with less tau. This is the practical version of the intuition, and it is what makes measuring tau before choosing therapy matter.
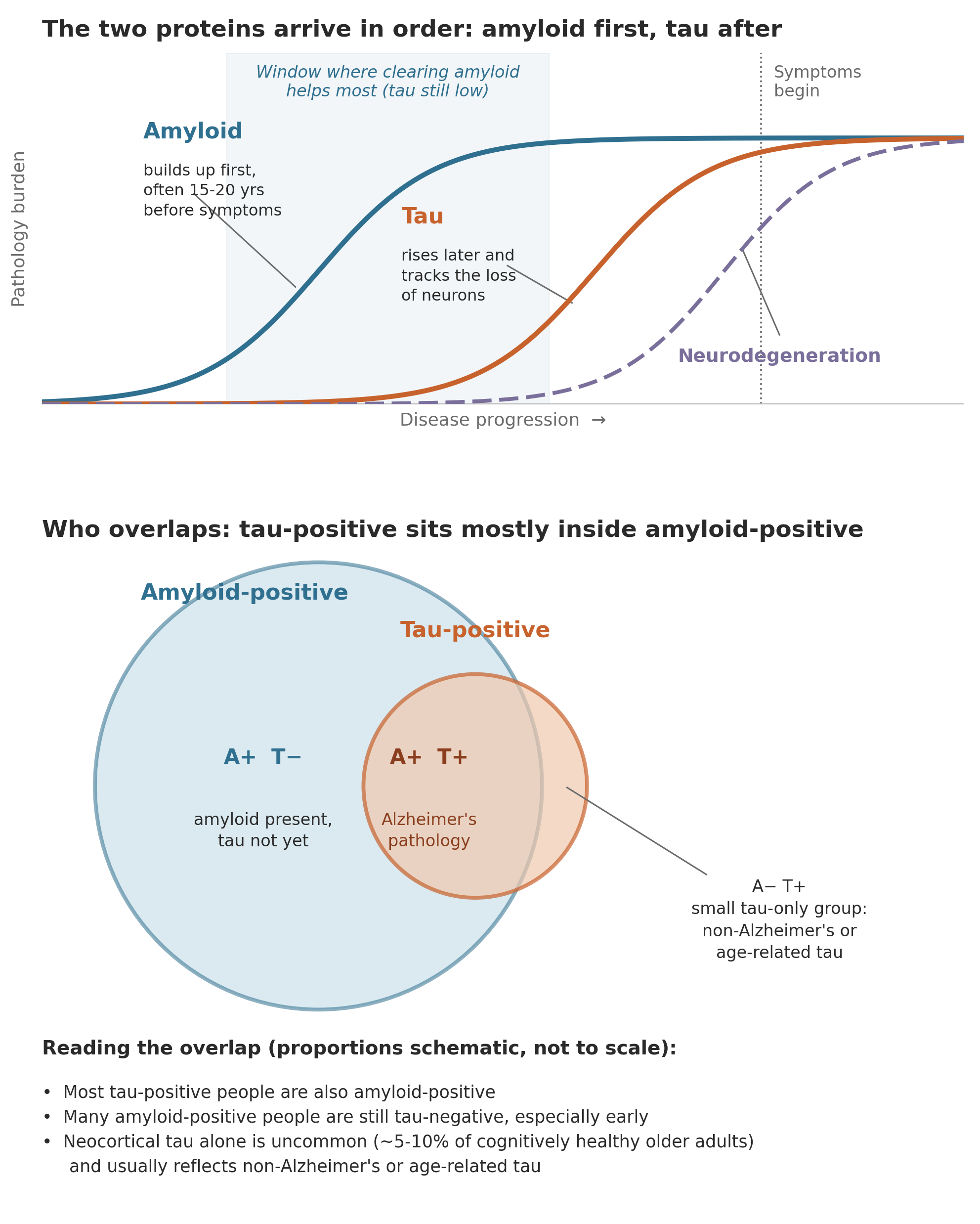
Donanemab shows this directly, because its pivotal trial was designed around it. In TRAILBLAZER-ALZ 2, participants were sorted by tau on PET imaging into a low-to-medium tau group and a high tau group, the latter hypothesized to be harder to treat because it reflects more advanced disease. In the low-to-medium tau group, donanemab slowed decline by about 35 percent on the trial’s main cognitive-functional scale. In the high tau group, the benefit was smaller and less consistent across measures. A 2026 post-hoc analysis reinforced the pattern, finding the greatest effect in people treated at an earlier disease stage, whether stage was defined by tau PET or by blood p-tau217.
Lecanemab points the same way, if less starkly. In the Clarity AD tau imaging substudy, benefit appeared across tau levels, but the highest rates of stability and improvement were in participants who began with little or no tau. Taken together, the two drugs support a simple clinical read, which is that clearing amyloid does the most good before tau has built up, and does progressively less once it has. That is the argument for knowing a person’s tau status, and for adding a tau-directed therapy in people whose tau is already high enough that amyloid clearance alone tends to underperform.
A blood test can now show how much of each pathology a person carries and roughly where they sit in the disease
Until recently, sorting people by tau meant a PET scan, which is expensive and available in few places. That is the barrier blood markers are removing, by capturing both proteins from an ordinary blood draw and making the amyloid-versus-tau distinction usable in an ordinary clinic rather than only in a trial.
Two blood measures do most of this work. The first is p-tau217, a form of tau that rises early, around the time amyloid becomes detectable, and that tracks amyloid pathology closely enough to identify it from an ordinary blood draw. The second is a newer marker called eMTBR-tau243, which reflects aggregated tau specifically and rises later, as tangles accumulate. A study in JAMA Neurology combined the two into a plasma staging model that matched PET-based staging closely, with agreement around 0.91 on a standard concordance measure and strong alignment with autopsy-confirmed pathology. A review in The Lancet Neurology lays out how p-tau217 is strong for detecting amyloid but less precise for tau staging, which is where the tau-specific marker adds information.
Blood measures can also estimate timing, not just presence. A model published in Nature Medicine used repeated p-tau217 measurements to estimate the age at which a person’s markers crossed into positivity, and from that estimated when symptoms were likely to begin, within a margin of about three to four years in research cohorts. I covered that work in The p-tau217 Alzheimer’s clock can now estimate when symptoms are likely to begin. These tools are built in research populations and are not yet meant to hand any single person a personal date, but they demonstrate that the information needed to place someone on the disease timeline is sitting in a blood sample.
What combination therapy could look like when matched to a blood profile
Put the pieces together and a workflow starts to take shape. A person whose blood shows amyloid but little aggregated tau sits where clearing amyloid has its strongest benefit, and where a subcutaneous option now makes starting treatment far less demanding. A person whose blood shows amyloid alongside high tau sits where the trials say amyloid clearance alone does less, which is exactly the situation where adding a tau-lowering drug has a rationale rather than clearing amyloid and hoping. That second group is the one diranersen speaks to, since its whole purpose is to lower the tau that appears to blunt the amyloid drugs once it accumulates. And because these markers can be measured repeatedly, the same blood draw that guides the choice can track whether a therapy is doing what it is meant to, adjusting the regimen over time rather than fixing it at the start. Trials testing amyloid-plus-something combinations are already registered, and the diranersen data give the tau half of that equation its first real clinical footing.
This is the precision-medicine model the ADDF has described, where roughly 75 percent of the current pipeline targets pathways beyond amyloid. Inflammation, metabolism, and neurotransmitter systems all sit in that non-amyloid share, which means the eventual combinations may reach past the two signature proteins entirely. The two announcements this week are early markers of that direction, one adding a target and one making an existing target easier to reach.
The limits that still apply
None of this is a cure, and the effect sizes describe slowing, not reversal. Diranersen still has to confirm its benefit in phase 3, and the unresolved dose question means the optimal way to use it is not yet known. Its delivery into spinal fluid is more involved than a subcutaneous shot. Blood-based tau staging is less precise at separating tau stages than it is at flagging amyloid, so it complements imaging rather than fully replacing it for now. The amyloid antibodies still carry the imaging-abnormality risk that requires monitoring, at home or not. What has changed is the shape of the plan. For the first time there is a plausible route to measuring which pathology a person carries, choosing therapies to match, giving at least some of them at home, and remeasuring to see whether the choice was right, which is closer to how the rest of medicine already treats complex chronic disease.
The longer arc points toward prevention matched to a person’s genetics
Further out, the direction all of this points is toward using these drugs before symptoms rather than after. If a blood test can estimate the years a person has before decline begins, the same test could mark the moment to start a low-burden therapy, an at-home injection that clears amyloid while tau is still low and the drug works at its best, which moves treatment closer to prevention. Matched to a person’s blood profile, that early regimen would not be one drug for everyone. Amyloid and tau are the two proteins with therapies in hand, but they are not the whole genetic picture of who develops Alzheimer’s. APOE remains the strongest common risk factor for the ordinary late-onset form, though it is one contributor among many rather than the whole story. Genome-wide studies have now tied roughly 75 regions of the genome to Alzheimer’s risk, and the combined effect of those common variants can be summed into a polygenic risk score that reaches well beyond APOE and the rarer immune-gene changes like TREM2. Many of the variants cluster in the immune and microglial machinery and in how brain cells handle lipids and move material internally, which points to whole biological pathways a future drug might act on rather than any single gene.
Underneath all of this is the recognition that Alzheimer’s is not one disease. Cerebrospinal fluid studies have begun to sort it into several molecular forms with distinct genetic profiles, differing in the biology that drives them and in how fast they progress. The eventual goal is therefore less a single treatment than a way to match each person’s biological form and genetic profile to the drug or combination most likely to help them. That is a long way off, and it will take drugs aimed at each of these forms before matching becomes possible. The two reports this week are a few small steps toward it, one showing that a tau-lowering drug can slow decline and one making an amyloid drug easier to give and to combine. A future in which a blood draw sets a person’s profile and a tailored combination is chosen to fit it remains years of trials away, though each of the pieces needed to build it is now visible rather than hypothetical.

Written by
Dr. Christin Glorioso, MD PhD
Dr. Glorioso is the founder and CEO of NeuroAge Therapeutics. With her background in neuroscience and medicine, she is dedicated to revolutionizing brain health and helping people maintain cognitive vitality.
Learn more about Dr. Glorioso

