
The Alzheimer’s Pipeline Is Finally Catching Up to the Biology
Why we need to shift our horizon, the most promising clinical trials, and how to enroll in them

People ask me all the time whether there is anything real coming for Alzheimer’s prevention and treatment, and whether they should be hopeful. My answer has shifted in the past three years from cautiously optimistic to pretty certain that we are riding a new wave that will yield real solutions. Here I explain why.
The history of Alzheimer’s drug development is, by any measure, one of the most challenging records in medicine. A comprehensive analysis of clinical trials from 2002 to 2012 found a 99.6% failure rate across all drug candidates, compared to 81% for cancer. (Cummings et al. 2014) More recent reviews put the Phase 2 and Phase 3 success rate since 2003 at around 2%, even counting the controversial 2021 aducanumab approval. (Aggarwal et al. 2022) For decades, every promising mechanism would work beautifully in mice and then collapse in humans. The field became synonymous with expensive, high-confidence failure.
Part of what drove those failures was a fundamental mischaracterization of what Alzheimer’s disease actually is. For most of the past three decades, Alzheimer’s was treated as a single disease with a single cause, as though amyloid plaques accumulate, neurons die, and dementia follows. Drug after drug was designed to interrupt that one mechanism, and most failed. But the more we have studied the biology at scale, using transcriptomics, proteomics, genetics, and imaging across thousands of people, the clearer it has become that Alzheimer’s is not one disease any more than cancer is one disease. Lung cancer and leukemia share a name and a cellular origin story, but they require completely different treatments targeting completely different biology. Alzheimer’s is similar. One person’s dementia may be driven predominantly by vascular injury and white matter disease, another’s by relentless amyloid accumulation amplified by APOE4 lipid dysregulation, a third’s by chronic neuroinflammation rooted in microglial dysfunction, and a fourth’s by mitochondrial dysfunction driving bioenergetic failure in neurons. These are overlapping but distinct biological trajectories converging on a shared clinical syndrome. Treating all of them with the same anti-amyloid antibody is roughly analogous to treating every cancer with the same chemotherapy.
This shift in understanding is what makes the current moment feel categorically different from what came before. The field is diversifying beyond amyloid to target neuroinflammation, tau biology, vascular health, metabolic pathways, senescence, and the underlying program of brain aging itself. And critically, it is beginning to use biomarkers to match the right intervention to the right patient rather than enrolling heterogeneous populations and averaging away the signal.
The next step, and the one that will define the next decade of Alzheimer’s medicine, is using artificial intelligence to make that matching systematic, directing the right drug to the right biological subtype before Phase 3, rather than learning that lesson through expensive late-stage failures.
There are currently 138 novel drugs in 182 active clinical trials across more than 4,500 sites worldwide. (Cummings et al. 2025) The target categories have diversified beyond amyloid. And the field is beginning to understand a pattern of failure that clarifies exactly where those drugs will need to be deployed to succeed.
The pattern is that almost every intervention that works in prevention or in early biology fails when tested in patients who already have symptomatic disease with confirmed pathology, and those failures are not random. They are telling us something about the window of opportunity for effective treatment, and about who needs to be identified and reached before that window closes.
Where the pipeline stands right now
The therapeutic landscape has shifted from a nearly exclusive focus on amyloid removal to a broader multi-target framework. Here is a summary of the current approved therapies and the most active Phase 3 agents.
One of the most consequential near term shifts in the pipeline is the emergence of oral small molecules targeting amyloid biology. The approved antibodies lecanemab and donanemab require IV infusions every two to four weeks at specialized infusion centers, plus periodic MRI monitoring for ARIA, which effectively restricts treatment to patients who live near academic medical centers and can commit to a months-long infusion schedule. For truly presymptomatic individuals who feel healthy, that burden alone is enough to prevent participation. AR1001, buntanetap, and valiltramiprosate are all daily pills taken at home, which dramatically improves access. Widening geographic reach and reducing patient burden are prerequisites for treating Alzheimer’s at the population scale the disease demands.
Table 1. FDA-Approved Disease-Modifying Therapies for Alzheimer’s Disease (as of 2025)
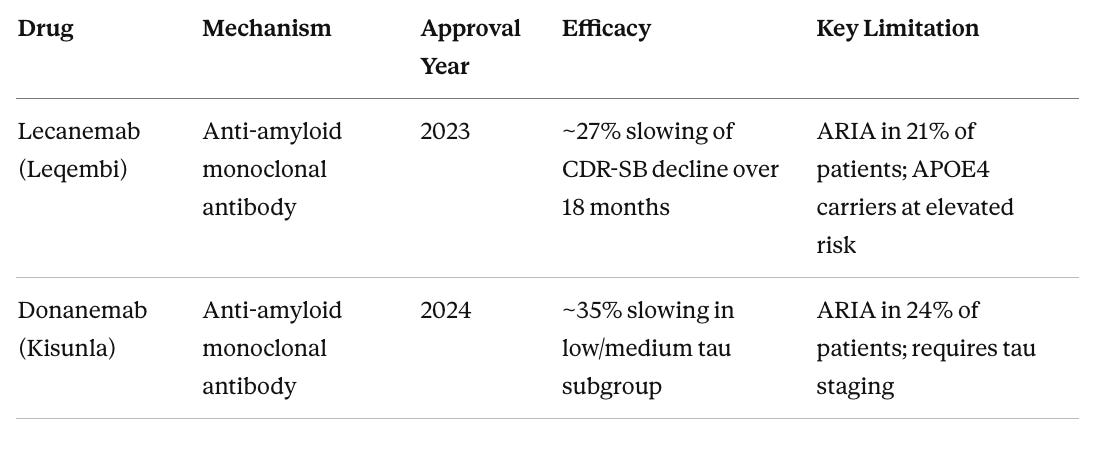
ARIA (amyloid-related imaging abnormalities) refers to temporary brain swelling or microbleeds that require MRI monitoring. CDR-SB (Clinical Dementia Rating Sum of Boxes) is the standard clinical endpoint measuring cognition and daily function across six domains.
Both approvals are meaningful proof of concept that removing amyloid from the brain can influence disease trajectory. But the effect sizes are modest, the patient populations are narrow (early symptomatic disease with confirmed amyloid), and APOE4 carriers face a troubling catch in that those at highest genetic risk may be least suitable for the currently available treatments due to elevated ARIA risk.
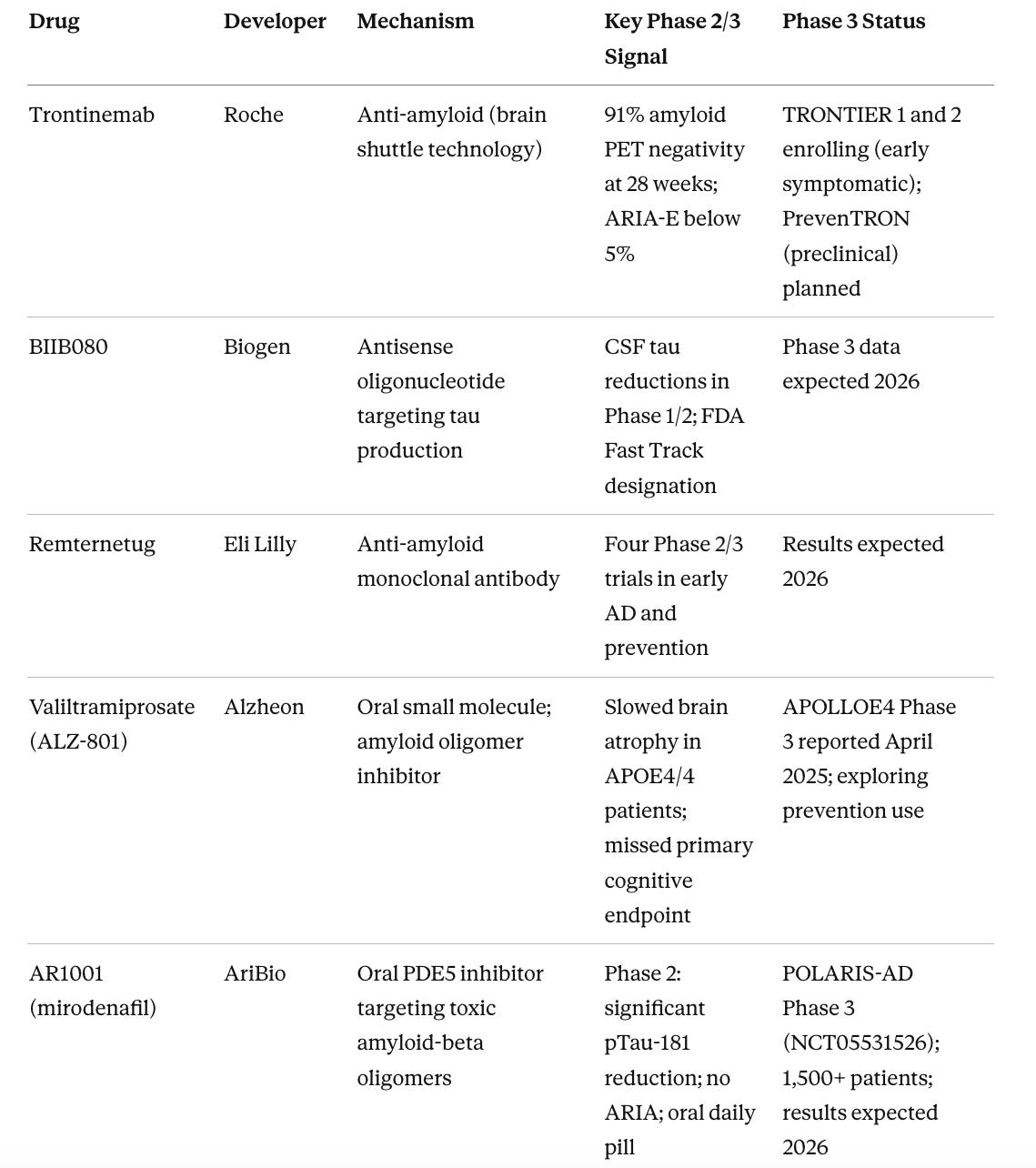
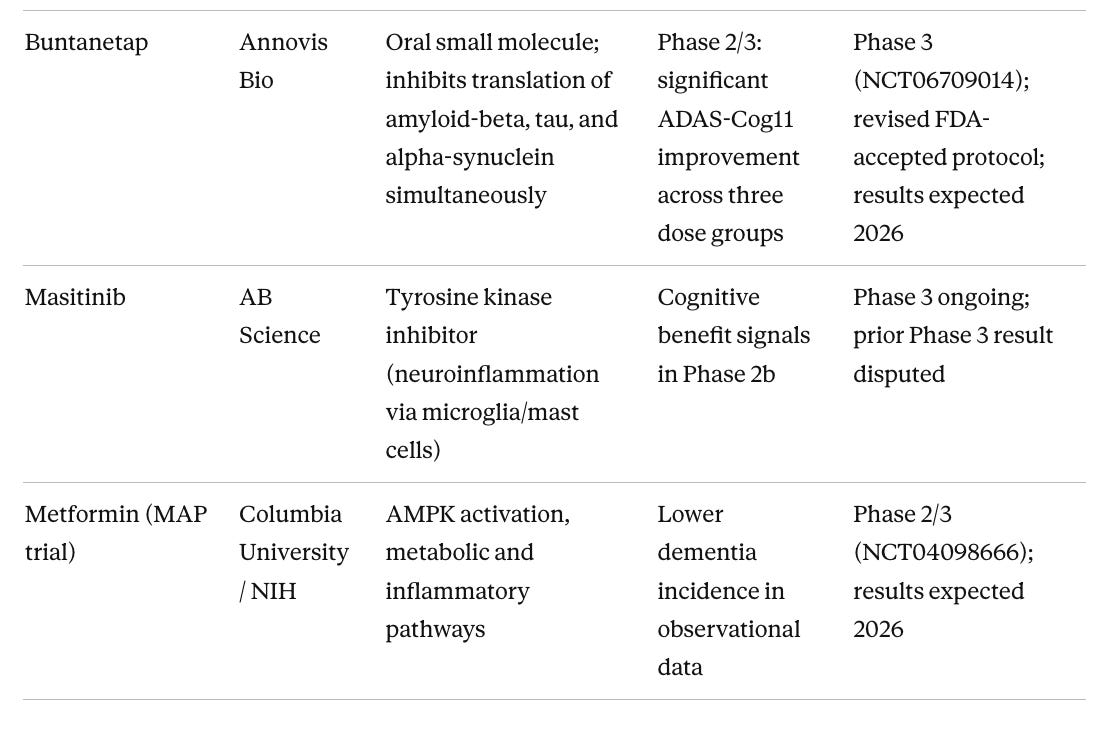
Table 2. Phase 3 Pipeline: Most Advanced Non-Approved Candidates
A note on metformin
Metformin gets more attention in longevity circles than its evidence base warrants. The much-discussed TAME trial, designed to test metformin against aging itself as a composite endpoint across cardiovascular disease, cancer, dementia, and all-cause mortality, has not launched after a decade of trying, due to persistent underfunding. The FDA agreed to the trial design back in 2015, and that regulatory template has real value, but the trial itself does not exist. What does exist is the MAP trial, a separate Phase 2/3 study at Columbia running in 326 people with amnestic MCI, with results expected in 2026. (Luchsinger et al. 2025) There are also unresolved concerns about metformin and Parkinson’s disease risk. Metformin inhibits mitochondrial complex I, the same pathway disrupted by the toxins rotenone and MPTP, a concern I cover in more depth in my Substack. The MAP results will be informative, but I would not recommend metformin off-label for brain health in healthy non-diabetic people in the meantime.
Table 3. Neuroinflammation and Aging-Targeted Approaches in Earlier Phases
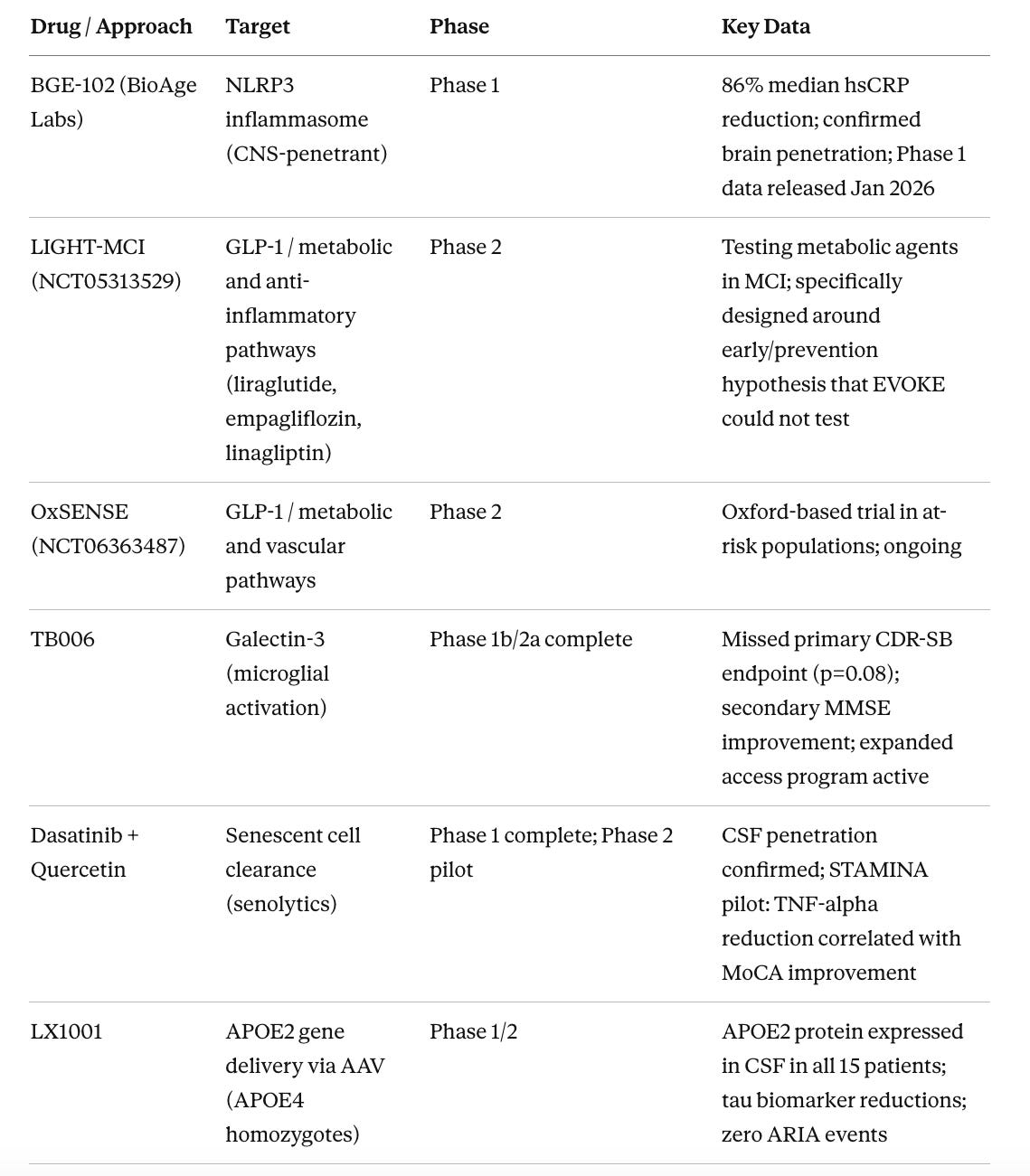
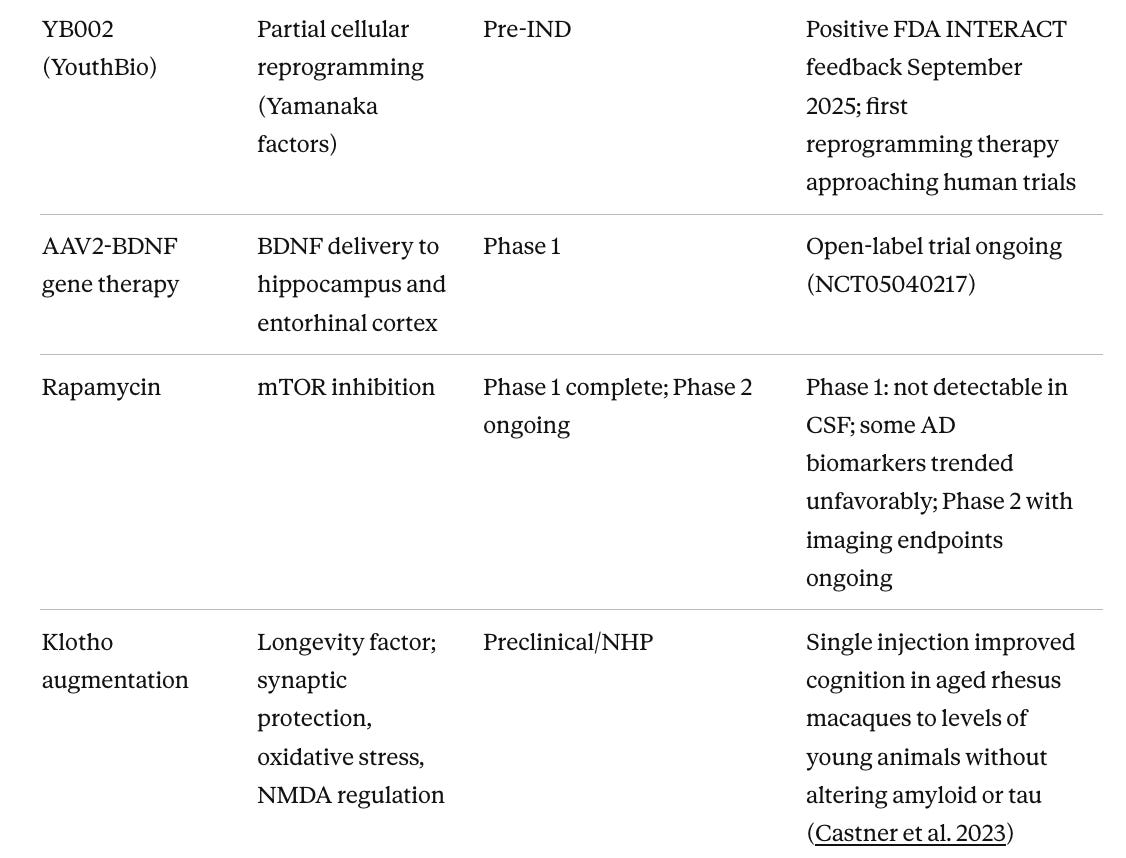
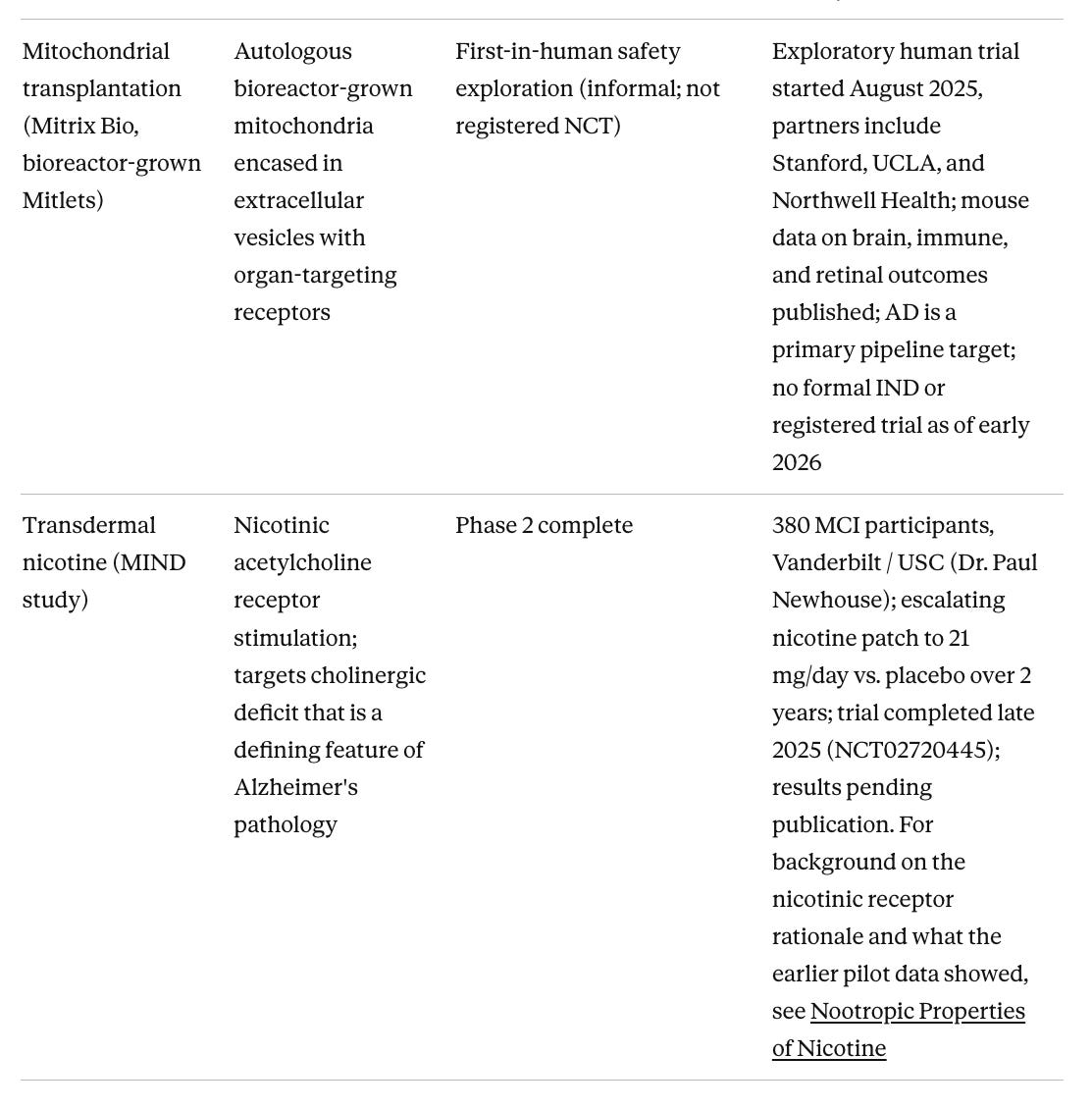
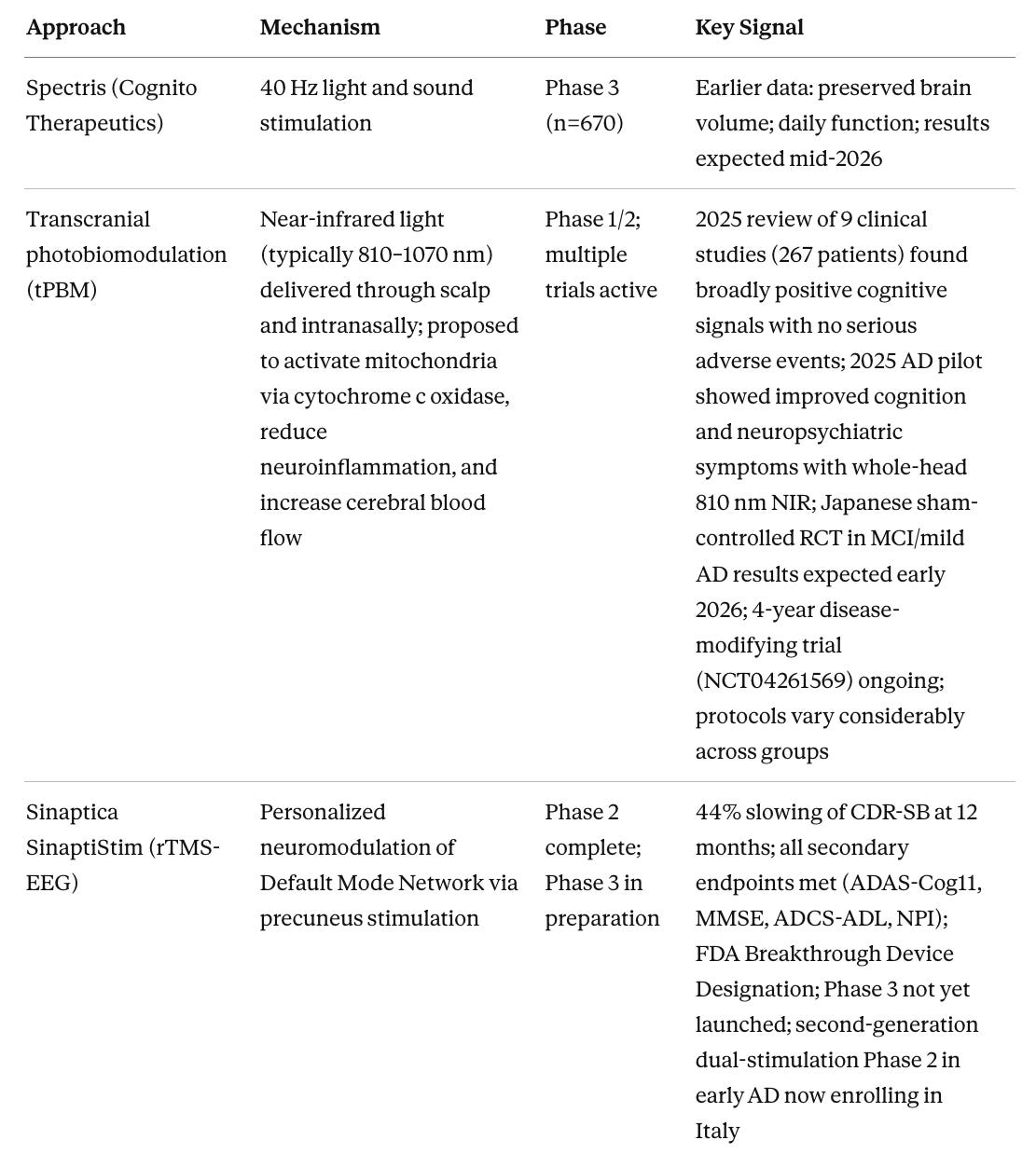
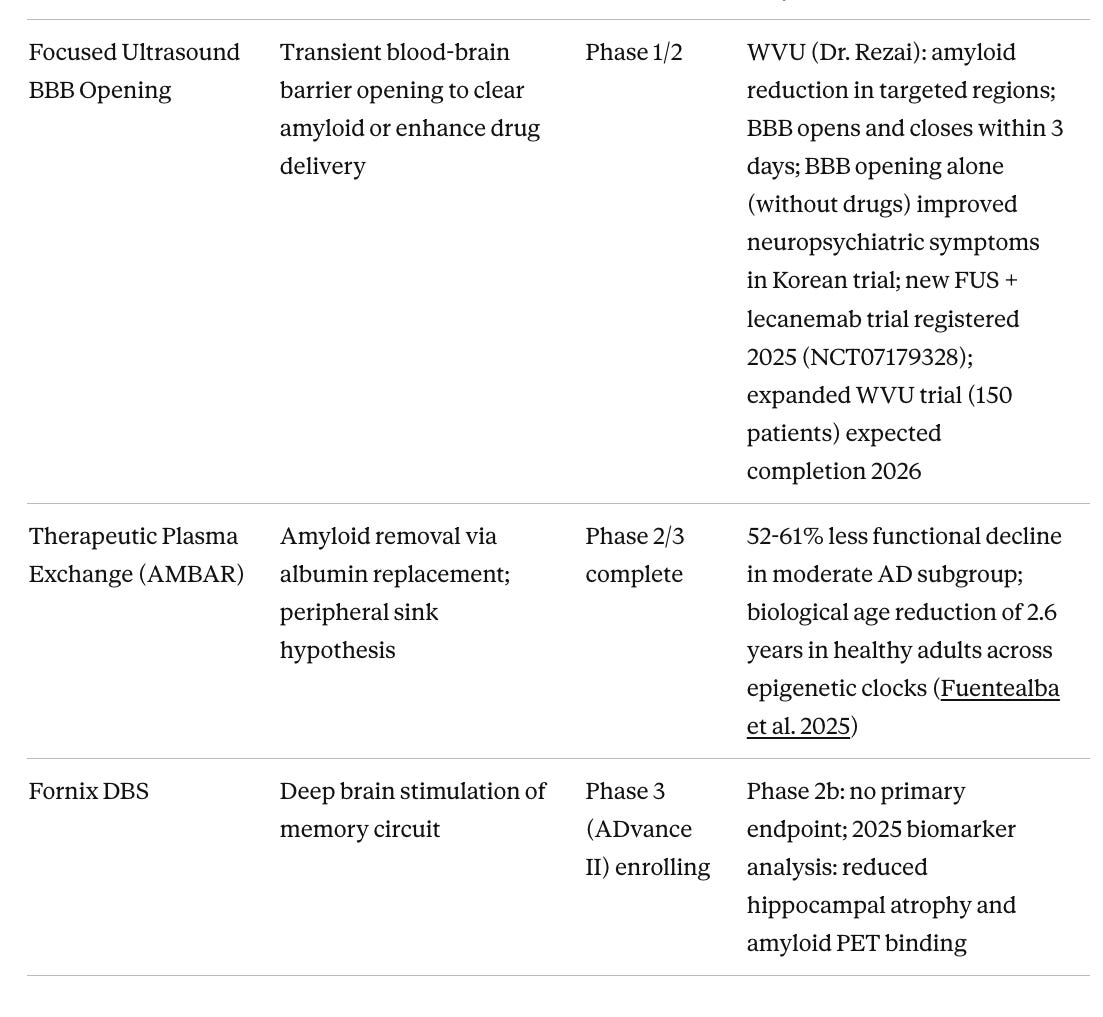
Table 4. Device-Based and Non-Pharmacological Approaches
The EVOKE trial and what GLP-1 results are telling us
The EVOKE and EVOKE+ trials of oral semaglutide enrolled 3,808 people with amyloid-confirmed MCI or mild dementia and followed them for two years. The results were negative across every cognitive and functional outcome. (Cummings et al. 2026, The Lancet) This was not entirely surprising, given that people taking GLP-1 agonists for diabetes or obesity show substantially lower rates of Alzheimer’s diagnosis in real-world data. One analysis found a hazard ratio of 0.33 for incident AD in semaglutide users versus insulin users (Nørgaard et al. 2022) but those signals come from prevention contexts over years, not from treating established disease. GLP-1 agonists likely work by modifying the metabolic and inflammatory substrate before pathology consolidates. By the time someone has confirmed amyloid-positive symptomatic AD, that window has passed. Semaglutide did improve CSF biomarkers by around 10%, suggesting the biology is real. It just cannot reverse established disease in two years, any more than lowering blood pressure reverses decades of arteriosclerosis.
The GLP-1 story is not closed. The liraglutide ELAD trial also missed its primary endpoint, but predefined secondary outcomes showed nearly 50% less brain volume loss and 18% slower cognitive decline, signals built into the trial design, not post-hoc, that raise legitimate questions about whether FDG-PET metabolic rate was the right primary endpoint for a drug acting through trophic and inflammatory mechanisms. (Edison et al. 2025) Two ongoing trials are testing the prevention hypothesis directly: LIGHT-MCI (NCT05313529) and OxSENSE (NCT06363487), both designed specifically for the earlier population that EVOKE could not address. Whether injectable GLP-1 formulations with higher CNS bioavailability, or combination approaches layered on top of amyloid clearance, produce different results remains an open and scientifically legitimate question.
Why this pattern appears across the pipeline
The GLP-1 story is one prominent example of a broader pattern in Alzheimer’s drug development. Consider the arc of the anti-amyloid antibody program. Bapineuzumab, solanezumab, crenezumab, and gantenerumab all failed in symptomatic disease populations across Phase 3. Most removed amyloid biologically but produced no cognitive benefit. The field spent roughly fifteen years arguing about whether the amyloid hypothesis was wrong. It turned out the hypothesis was partially right, but we need additional approaches and the timing was wrong. Lecanemab and donanemab succeeded not because they are fundamentally different antibodies, but because they were tested in earlier disease (MCI and mild dementia with confirmed amyloid rather than moderate dementia), with better biomarker-based patient selection, and with more sensitive clinical endpoints.
The same timing logic applies to the failed gamma-secretase and beta-secretase inhibitors, to the early tau aggregation inhibitors, and now almost certainly to the GLP-1 agonists. These are not random failures of pharmacology but are instead informative failures about when in the disease course intervention is possible.
By the time a patient develops symptomatic Alzheimer’s disease, amyloid deposition has been accumulating for 15 to 20 years. Tau pathology has been propagating through the brain for a decade, synaptic density in the hippocampus and entorhinal cortex has declined substantially, and neuroinflammation has been chronically active. An intervention that works by slowing one of those upstream processes, whether it reduces amyloid production, dampens inflammation, or restores metabolic resilience, is being asked to reverse a decade or two of accumulated damage in the space of an 18-month trial. The effect sizes are small not because the mechanisms are wrong but because the downstream pathology has its own momentum.
This is why the drugs with the best near-term prospects in treatment settings are those that target the most proximal pathology in already-symptomatic patients. Trontinemab’s brain shuttle technology matters because it achieves far higher amyloid clearance faster than existing antibodies, with lower ARIA risk, which may extend the treatment window. BIIB080’s antisense approach to tau matters because tau burden correlates more tightly with cognitive symptoms than amyloid does, and targeting tau production directly rather than clearing extracellular aggregates is more mechanistically proximal. But even these agents will likely show their strongest benefit in patients identified before substantial synaptic loss has occurred.
The approaches most likely to succeed and why
Based on convergent evidence across preclinical, NHP, and early human data, here is my assessment of where the evidence is strongest.
Trontinemab has the most compelling near-term profile in the treatment category. The mechanism is proven. Lecanemab and donanemab already established that amyloid clearance influences disease trajectory. Trontinemab’s advantage is in delivery. Its brain shuttle technology achieved 91% amyloid PET negativity at 28 weeks, compared to roughly 70% for lecanemab at 18 months, with ARIA-E rates below 5% versus 13-24% for approved therapies. For APOE4 homozygotes, who currently have almost no treatment options due to elevated ARIA risk, this could be the first viable anti-amyloid option.
BIIB080 combined with an amyloid-clearing agent represents the most mechanistically coherent combination logic in the field. Clearing amyloid first with a brain-shuttle antibody, then suppressing tau propagation with an antisense oligonucleotide, targets the two defining pathologies sequentially in the order they become clinically relevant. The Phase 1/2 tau reduction data from BIIB080 support the mechanism. Full Phase 3 data are expected in 2026.
AR1001 (mirodenafil) warrants attention because it is an oral daily pill, not an IV infusion requiring clinic visits and MRI monitoring. The approved anti-amyloid antibodies are large proteins that cannot survive the digestive tract, so they must be delivered intravenously, which means infusion centers, two-to-four-week schedules, and the ARIA monitoring infrastructure that goes with them. AR1001 is a small molecule that crosses the gut wall and enters the bloodstream like any other pill. In Phase 2 it produced significant reductions in plasma pTau-181 with no ARIA events. If those results hold in the 1,500-patient POLARIS-AD Phase 3 trial, the practical implications extend well beyond the drug itself. An oral anti-amyloid therapy that can be prescribed in primary care and taken at home could reach patients who currently never access treatment because they live too far from an infusion center, cannot commit to monthly clinic visits, or are in a stage of disease where prevention feels premature but biology says otherwise.
Buntanetap (Annovis Bio) is the only drug in Phase 3 targeting amyloid-beta, tau, and alpha-synuclein translation simultaneously via a single oral small molecule. The Phase 2/3 dose-ranging study showed statistically significant ADAS-Cog improvements across three dose groups, which is a cleaner signal than most oral small molecule programs have produced. It reduces synthesis of all three aggregating proteins at the ribosomal level rather than targeting the aggregates themselves, an upstream approach that is disease-agnostic across multiple neurodegenerative conditions. Results in 2026 will be important for understanding whether the multi-target oral approach can replicate in a larger population.
LX1001 (APOE2 gene therapy) targets the single most consequential genetic risk factor in Alzheimer’s from a different direction. Rather than inhibiting APOE4 directly, it delivers the protective APOE2 allele via an AAV vector directly to the CSF, specifically in APOE4 homozygotes who carry the highest genetic risk and are excluded from most anti-amyloid trials due to ARIA. In Phase 1/2, APOE2 protein was detectable in the CSF of all 15 patients, tau biomarkers trended downward, and there were zero ARIA events. The early human data are credible, and if the Phase 2 results confirm the biomarker signal alongside a cognitive trend, LX1001 could represent the first precision genetic therapy for Alzheimer’s in the highest-risk group.
Klotho augmentation has arguably the most compelling NHP data of anything in the pipeline. Dr. Castner and colleagues showed in 2023 that a single peripheral injection of klotho protein improved cognition in aged rhesus macaques to levels indistinguishable from young animals, without altering amyloid or tau at all. (Castner et al. 2023) This suggests that restoring endogenous brain resilience is sufficient to reverse age-related cognitive decline independently of classical AD pathology. The challenge is that no clinical-stage program has advanced this to Phase 2 in AD specifically, which represents an opportunity for the field.
BGE-102 (BioAge NLRP3 inhibitor) sits at a compelling mechanistic convergence point. NLRP3 is the inflammasome (a protein complex that initiates inflammatory signaling in immune cells) activated by amyloid aggregates in microglia, the brain’s resident immune cells. It drives neuroinflammation, propagates tau pathology, and operates behind the blood-brain barrier where peripheral anti-inflammatory drugs cannot reach. The January 2026 Phase 1 interim data showed 86% median hsCRP reduction with confirmed brain penetration. (BioAge Labs 2026) The full cognitive and biomarker outcomes in an AD population are not yet available, but the pharmacodynamic signal is clean.
Sinaptica’s personalized rTMS is a device program that I think has a good chance of demonstrating real efficacy at scale. I have written about TMS at length on my Substack. The Phase 2 data show 44% slowing of CDR-SB over 12 months, with statistically significant improvements across cognition, daily function, and behavioral disturbances, and no serious side effects. The key distinction from earlier TMS programs is personalization, meaning calibration to each individual’s brain anatomy and connectivity using MRI, TMS-EEG, and neuronavigation, so that stimulation actually reaches the intended target. Phase 3 has not yet launched as of early 2026, but FDA Breakthrough Device Designation is in place and active preparations are underway. A second-generation system combining TMS with transcranial electrical stimulation is now being tested in early AD in Italy, with a 70% reduction in session time. To express interest in Sinaptica’s trial program, contact the company through sinapticatx.com or media contact Kathryn Morris at kathryn@brightpointny.com.
Focused ultrasound BBB opening may turn out to be important as the delivery technology that makes anti-amyloid antibodies work substantially better. Current approved antibodies penetrate the brain poorly, requiring high doses that drive ARIA risk. FUS transiently opens the blood-brain barrier for 48–72 hours with a non-invasive procedure, potentially allowing far lower antibody doses to achieve the same clearance. Dr. Ali Rezai’s program at West Virginia University has shown 14% amyloid reduction in targeted regions from BBB opening combined with aducanumab, a modest signal but from a trial not designed or powered to show cognitive change. The Korean trial of BBB opening alone, without any drug, and repeated bilateral frontal FUS improved neuropsychiatric symptoms, suggesting that clearing pro-inflammatory signals and toxic aggregates through mechanical BBB disruption may itself be therapeutic. A new trial combining FUS with lecanemab (NCT07179328) was registered in 2025 and represents the next logical test of whether the delivery enhancement hypothesis holds in a properly designed study. The WVU program at Rockefeller Neuroscience Institute is the most active US site, with contact information at wvumedicine.org/rockefeller-neuroscience-institute.
Partial cellular reprogramming has moved from preclinical promise to first-in-human testing faster than most of the field expected. Life Biosciences, co-founded by Dr. David Sinclair at Harvard, received FDA IND clearance in January 2026 and initiated Phase 1 of ER-100 (NCT07290244), a gene therapy using three Yamanaka factors (OCT4, SOX2, KLF4) to partially reprogram aged retinal cells, in patients with glaucoma and non-arteritic anterior ischemic optic neuropathy. This is the first human trial of partial epigenetic reprogramming anywhere. The first indication is the eye, which is a part of nervous system, and the same platform is being developed for neurodegeneration and other age-related diseases. For Alzheimer's specifically, YouthBio's YB002 received positive FDA INTERACT feedback in September 2025 and remains the most advanced reprogramming program targeting the brain, though it is still several years from a clinical trial. Mouse model data showing reversed epigenetic aging signatures, reduced amyloid plaques, and improved cognition are compelling, and Life Biosciences' human safety data from the eye trial will inform what is possible in the central nervous system.
Stem cell therapies represent a distinct category worth watching. I have covered the broader stem cell landscape for neurodegeneration in a recent Substack post, including why Parkinson's has been an easier target than Alzheimer's and what that means for the field. The most clinically advanced program is laromestrocel (Longeveron), a bone marrow-derived allogeneic MSC therapy whose Phase 2a RCT published in Nature Medicine in March 2025 met its primary safety endpoint and showed trends toward reduced brain atrophy and slowed cognitive decline at the highest dose. The drug appears to work via anti-inflammatory paracrine mechanisms rather than cell replacement. A separate Phase 1 of autologous adipose-derived stem cells (RB-ADSCs, Regeneration Biomedical) reported positive interim safety and early efficacy signals at ISCT 2025. iPSC-derived neural cell approaches, which would replace lost neurons rather than modulate inflammation, remain a longer horizon but are advancing. The appeal of the whole category is that it does not attempt to hit a single molecular target in a heterogeneous disease. Instead, it tries to restore the biological environment that allows neurons to survive.
The window problem and what it means for early detection
Across the entire pipeline, the clearest pattern in clinical trial outcomes is that interventions which modulate upstream biology, whether metabolic, inflammatory, vascular, or epigenetic, fail in symptomatic disease and show their strongest signals either in prevention populations or in the earliest identifiable disease stages. The anti-amyloid antibodies required a decade of Phase 3 failures before the field understood that patient selection and disease stage, not just mechanism, determined success. The GLP-1 agonists appear to be repeating that lesson at a faster pace.
This has a direct implication for how we should think about Alzheimer’s as a clinical problem. The biological processes that become Alzheimer’s disease are detectable long before symptoms appear. Transcriptomic signatures of brain aging shift measurably in the third decade of life. (Glorioso et al. 2011) Plasma proteomic brain age clocks predict dementia risk nearly 15 years before diagnosis. (Guo et al. 2024) Brain MRI volumetric patterns that predict conversion to dementia are visible in cognitively normal adults years before any clinical symptom. Amyloid accumulation begins 15-20 years before symptoms. Plasma biomarkers for phosphorylated tau (pTau217) now match CSF and PET accuracy for identifying amyloid pathology, making presymptomatic screening accessible in primary care settings.
The drugs most likely to succeed are not necessarily the ones that work against the most severe disease. They are the ones that work early, against the earliest detectable biology, in people who have not yet crossed the threshold into irreversible downstream pathology. That means identifying those people by having a reliable, accessible, and comprehensive assessment of biological brain age that can flag individuals who are aging faster than their chronological age suggests, who carry genetic risk factors that change their trajectory, and who have modifiable contributors to that risk that can be addressed before the window closes.
NeuroAge’s multi-modal assessment integrates MRI brain volumetrics, whole genome sequencing covering hundreds of genetic risk markers, RNA-based biomarkers, and cognitive performance testing into a unified picture of where someone’s brain is, how fast it is aging, and what that implies for their risk of cognitive decline over the coming decade or two. The drugs in Tables 2 and 3 are moving rapidly, and the patients who will benefit most are those identified early enough that those drugs can still work.
How to find and enroll in current trials
Clinical trial participation is one of the most direct ways to access cutting-edge therapies before approval and in many cases receive comprehensive biomarker assessments at no cost.
For people with MCI or early Alzheimer’s symptoms (confirmed amyloid)
TRONTIER 1 and TRONTIER 2, Roche’s Phase 3 trontinemab trials, are actively recruiting adults aged 50–90 with MCI or mild dementia and confirmed amyloid. APOE4 homozygotes are eligible, and prescreening starts with a blood-based pTau217 test rather than a PET scan. Find a site at genentech-clinicaltrials.com.
For people at risk but not yet symptomatic
TRAILRUNNER-ALZ 3 (Eli Lilly) is enrolling cognitively normal adults with positive plasma pTau217 to test remternetug as a prevention therapy via subcutaneous self-injection every three months. Site locator and contact are on the ClinicalTrials.gov page.
For people with a genetic Alzheimer’s mutation or strong family history
The DIAN-TU Primary Prevention Trial enrolls people from families with dominantly inherited early-onset AD mutations, beginning as early as 25 years before estimated symptom onset. Contact the Washington University team at dian@wustl.edu.
For healthy older adults interested in brain aging research
ADNI4 is a long-term observational study enrolling adults aged 55–90 for MRI, amyloid and tau PET, cognitive testing, and blood biomarkers. Not a drug trial, but one of the most important biomarker programs in the field, with both in-clinic and remote participation options.
For NeuroAge’s brain aging study
NeuroAge is currently recruiting people with mild cognitive impairment or early Alzheimer’s disease for a study evaluating our multi-modal brain aging assessment, with a personalized risk report provided to every participant. Our study is supported by funding from the Alzheimer’s Drug Discovery Foundation. Sign up here.
How to search more broadly
Alzheimer’s Association TrialMatch matches individuals to open trials by age, diagnosis, and location. Alzheimers.gov has plain-language summaries of eligibility. ClinicalTrials.gov is the complete registry. Search “Alzheimer’s disease” filtered by “Recruiting” and “Interventional.” Most significant trials require amyloid-positive status, and knowing your biomarker status before applying streamlines the process considerably.
The overall picture is that the field has moved from a single-target, late-stage, under-biomarkered approach to something substantially more sophisticated. The remaining challenge is identifying and reaching the right populations early enough that the drugs can still work.
Targeting aging itself is the path forward
The drugs most likely to produce the largest reductions in Alzheimer’s burden over the long run are probably not the ones targeting amyloid in 70-year-olds with established pathology. They are the ones that slow the rate of brain aging itself, including GLP-1 agonists used preventively in metabolically at-risk individuals, senolytics, mTOR modulators, klotho augmentation, and eventually partial epigenetic reprogramming. Every downstream pathology in this article (amyloid accumulation, tau propagation, microglial dysfunction, mitochondrial failure) becomes more tractable the earlier it is addressed, before decades of accumulated damage have their own momentum.
This means the next decade of Alzheimer’s medicine depends not just on better drugs, but on a clinical infrastructure for identifying who is at risk before symptoms appear. The measurement tools are maturing rapidly and are finally reaching the clinic, but they are not yet integrated into an assessment that tells a 50-year-old which aspect of their brain biology is most accelerated, which pathways are driving it, and which interventions are most likely to be effective for their particular profile.
AI will be central to closing that gap. Models trained on multi-modal biological data (brain imaging, plasma biomarkers, genomics, transcriptomics, cognitive trajectories) can already identify subgroups of individuals whose disease biology is distinct, predict who will respond to a given mechanism, and flag who is at elevated risk years before any clinical symptom appears. What has historically required the expensive failure of a 3,000-person randomized trial to learn may increasingly be answerable before Phase 3, by analyzing the molecular signatures of people who did and did not respond to a treatment and feeding that back into patient selection for the next trial.
NeuroAge was built around this gap. Our multi-modal assessment combines brain MRI volumetrics, whole genome sequencing covering hundreds of genetic risk markers, RNA biomarkers that track the transcriptional aging program identified in our foundational research, and cognitive performance testing into a unified biological brain age profile. The goal is not simply to tell someone their risk but to tell them which aspect of their biology is most accelerated, which modifiable factors are driving it, and which interventions are most likely to be effective for their specific profile. As the precision medicine framework for Alzheimer’s matures and AI-guided patient-treatment matching becomes routine, having that baseline biological characterization will determine who gets the right drug at the right time. Knowing where your brain is now is how you ensure you are in the right place when the drugs that actually work arrive.
If you want to participate in our study, you can sign up here.

Written by
Dr. Christin Glorioso, MD PhD
Dr. Glorioso is the founder and CEO of NeuroAge Therapeutics. With her background in neuroscience and medicine, she is dedicated to revolutionizing brain health and helping people maintain cognitive vitality.
Learn more about Dr. Glorioso

